4 Évaluation et exploration
4.1 Évaluation de la stabilité des clades
Il est utile d’évaluer la stabilité des clades obtenus. On peut le faire avec un ré-échantillonnage aléatoire avec remise (bootstrap) qui va simuler de la variation et permettre ainsi de quantifier la sensibilité des résultats aux données de l’échantillon observé. Cette méthode permet d’obtenir pour chaque branche de l’arbre la proportion d’arbres ré-échantillonnés qui contiennent cette branche et ainsi d’identifier les branches reposant sur un faible nombre de données.
On peut par exemple évaluer nos résultats obtenus par UPGMA.
Il faut au préalable récupérer la matrice de données. C’est totalement redondant mais il s’agit d’une contrainte technique. Il faut en plus définir une fonction qui sera appliquée à chaque échantillon, ici on utilise la composition de MatrixTophyDat(), Hamming() puis upgma(). On définit également à l’avance le nombre de ré-échantillonnages à effectuer, ici 1000.
lx_m <- PhyDatToMatrix(lx_phy)
boot_upgma_hamming <- function(x) {
upgma(Hamming(MatrixToPhyDat(x)))
}
n_bs <- 1000On peut alors lancer la procédure de ré-échantillonnage sur l’arbre obtenu par UPGMA et la matrice de données. Même avec 1000 ré-échantillonnages, on obtient les résultats en un temps très court.
La fonction set.seed() permet de fixer la graine aléatoire et d’assurer la reproductibilité des résultats.
set.seed(123456)
lx_upgma_bs <- boot.phylo(lx_upgma,
lx_m,
boot_upgma_hamming,
B = n_bs,
trees = TRUE,
quiet = TRUE
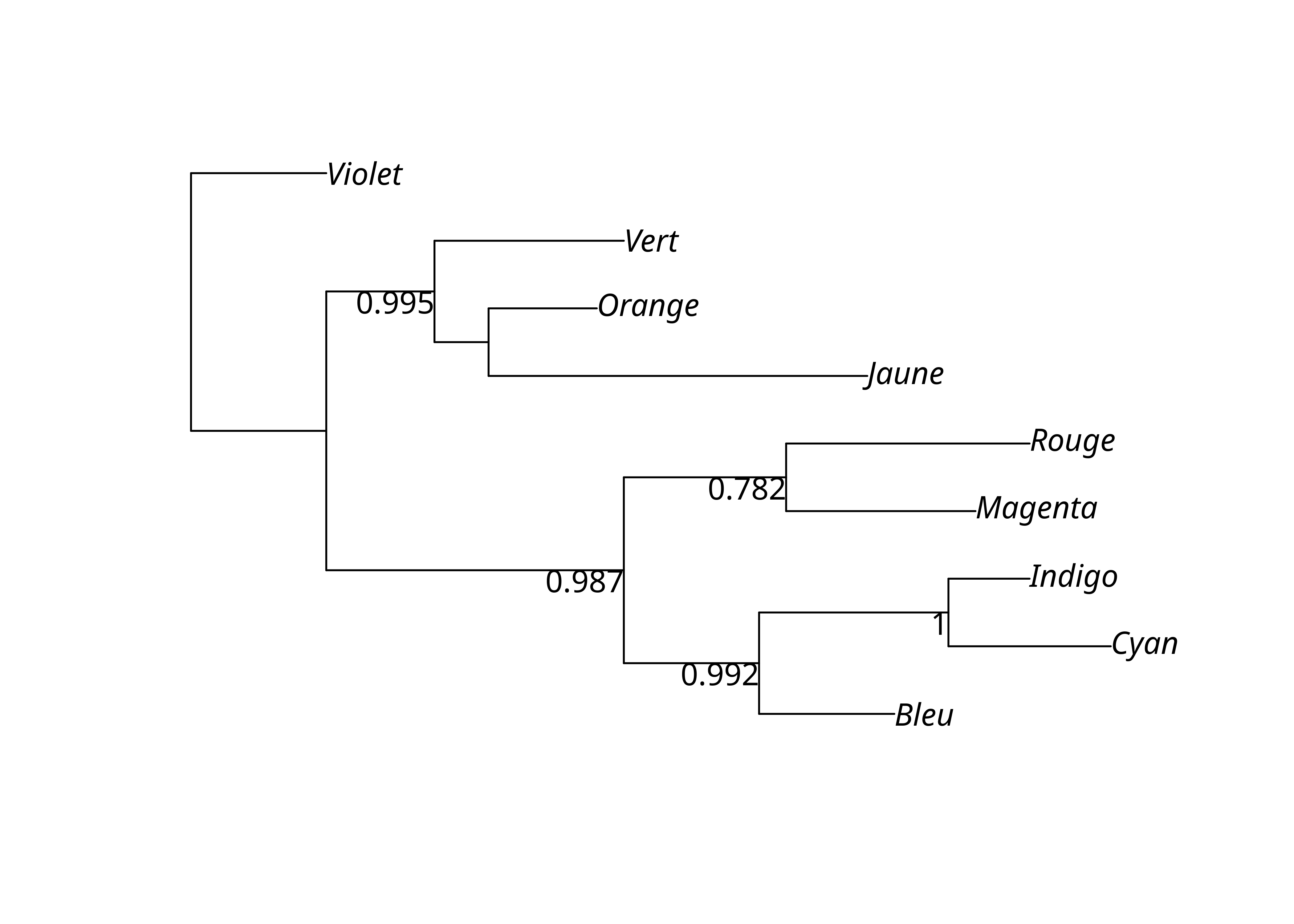
)On peut ainsi obtenir le nombre de ré-échantillonnages où chacun des clades de l’arbre par UPGMA apparaît, les transformer en proportions, et affecter ces valeurs aux nœuds internes de l’arbre. On peut alors les afficher sur le dendrogramme.
upgma_bs_scores <- prop.clades(lx_upgma, lx_upgma_bs$trees, rooted = TRUE)
upgma_support_pct <- upgma_bs_scores / n_bs
lx_upgma$node.label <- upgma_support_pct
plot(lx_upgma, show.node.label = TRUE)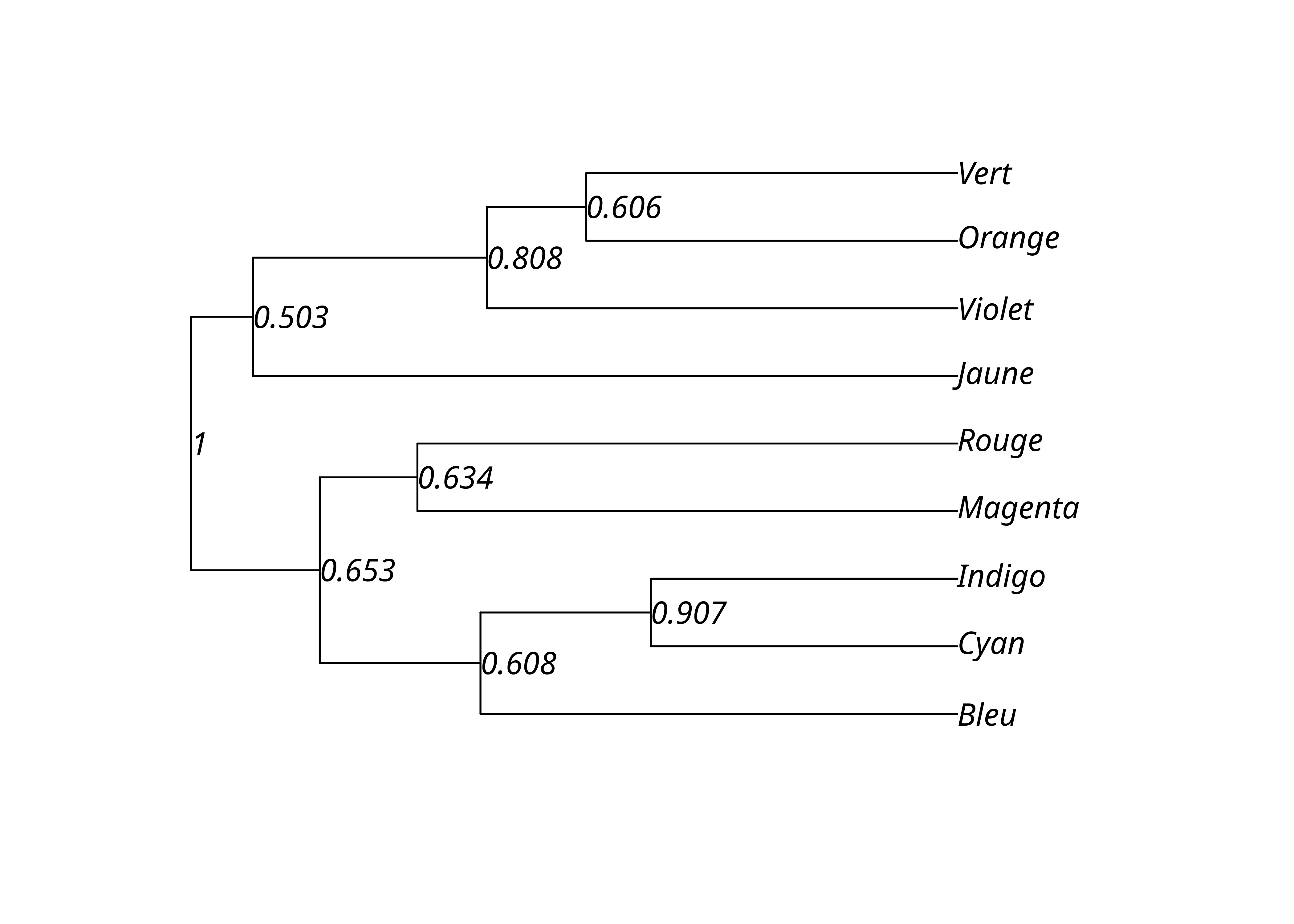
Une autre méthodes consiste à directement afficher les résultats avec plotBS().
plotBS(lx_upgma, lx_upgma_bs$trees)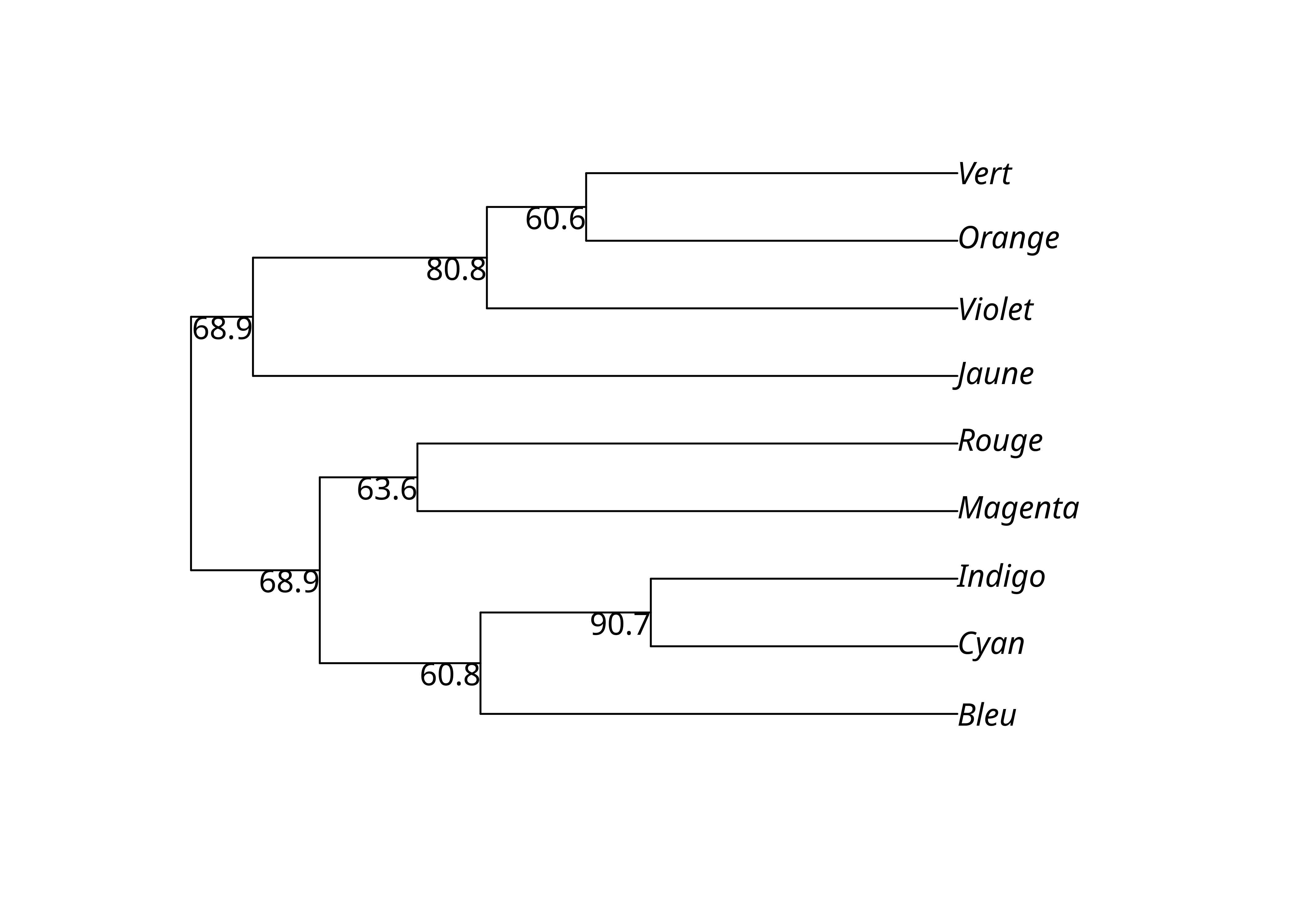
On peut choisir de n’afficher que les valeurs supérieures à un certain seuil, typiquement 70%.
upgma_support_pct[upgma_support_pct < .7] <- NA
lx_upgma$node.label <- upgma_support_pct
plot(lx_upgma, show.node.label = TRUE)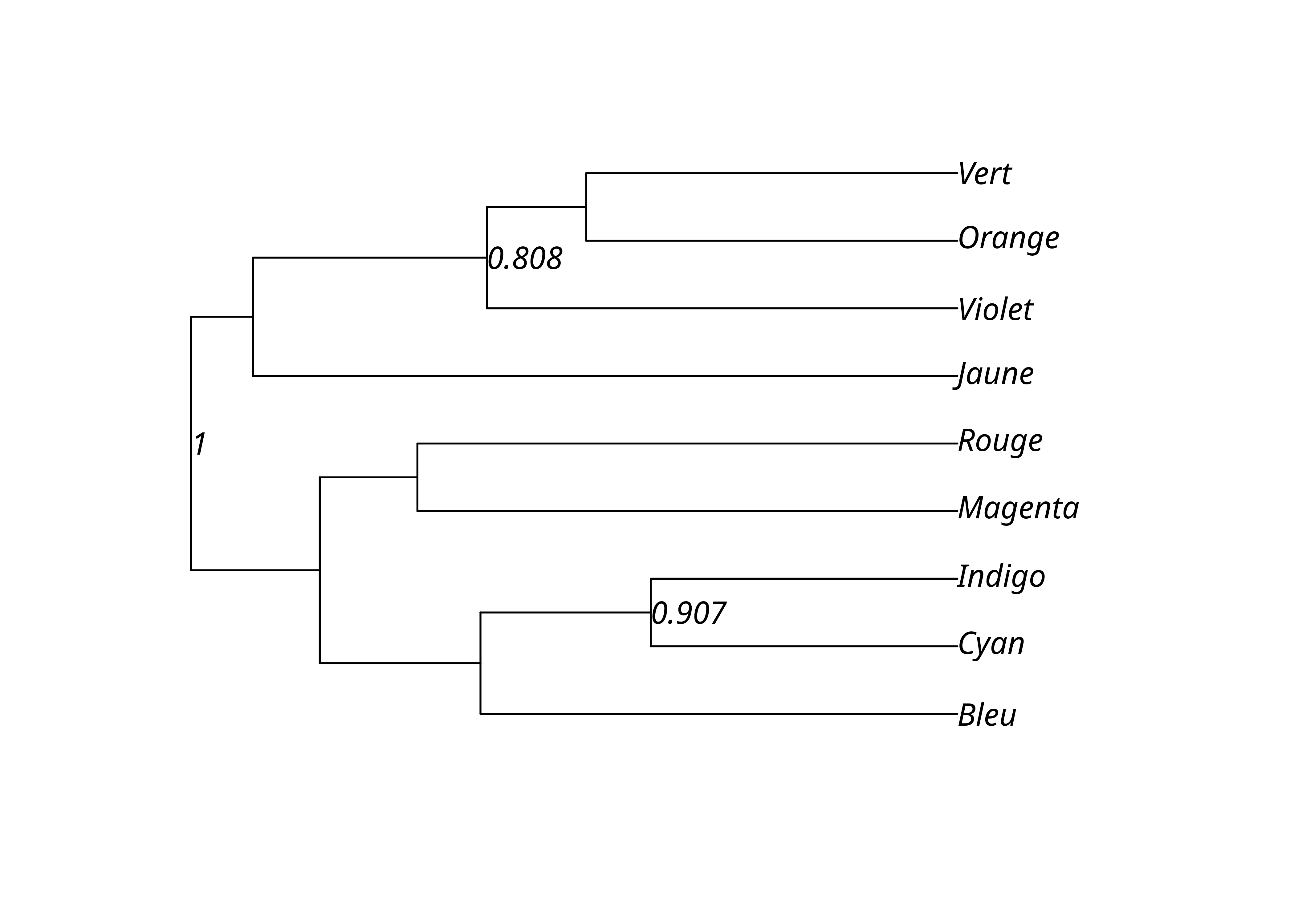
Ou plus simplement avec plotBS():
plotBS(lx_upgma, lx_upgma_bs$trees, p = 70)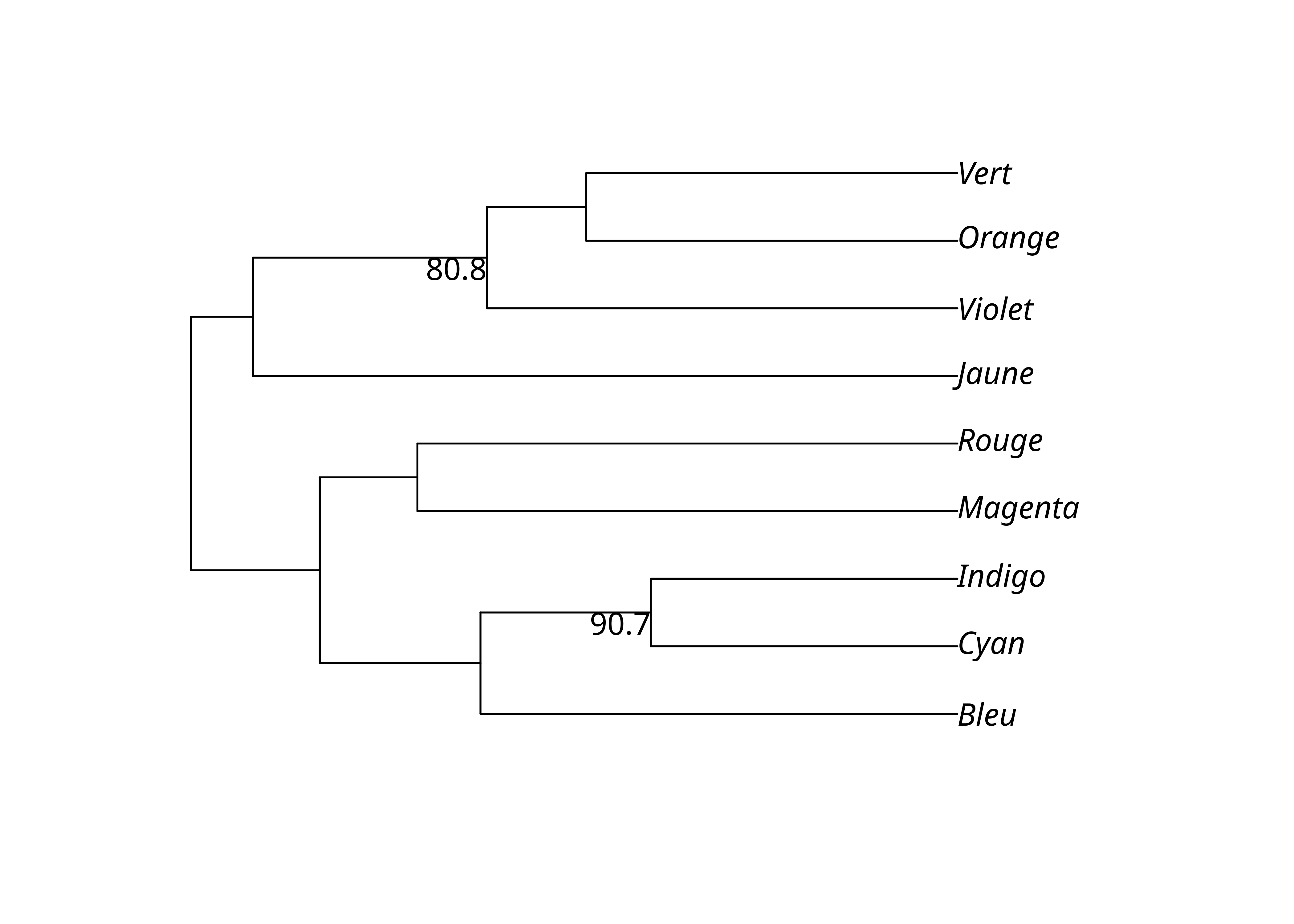
Un arbre parcimonieux obtenu avec pratchet() contient déjà des valeurs de bootstrap.
4.2 Résumer l’accord entre des résultats
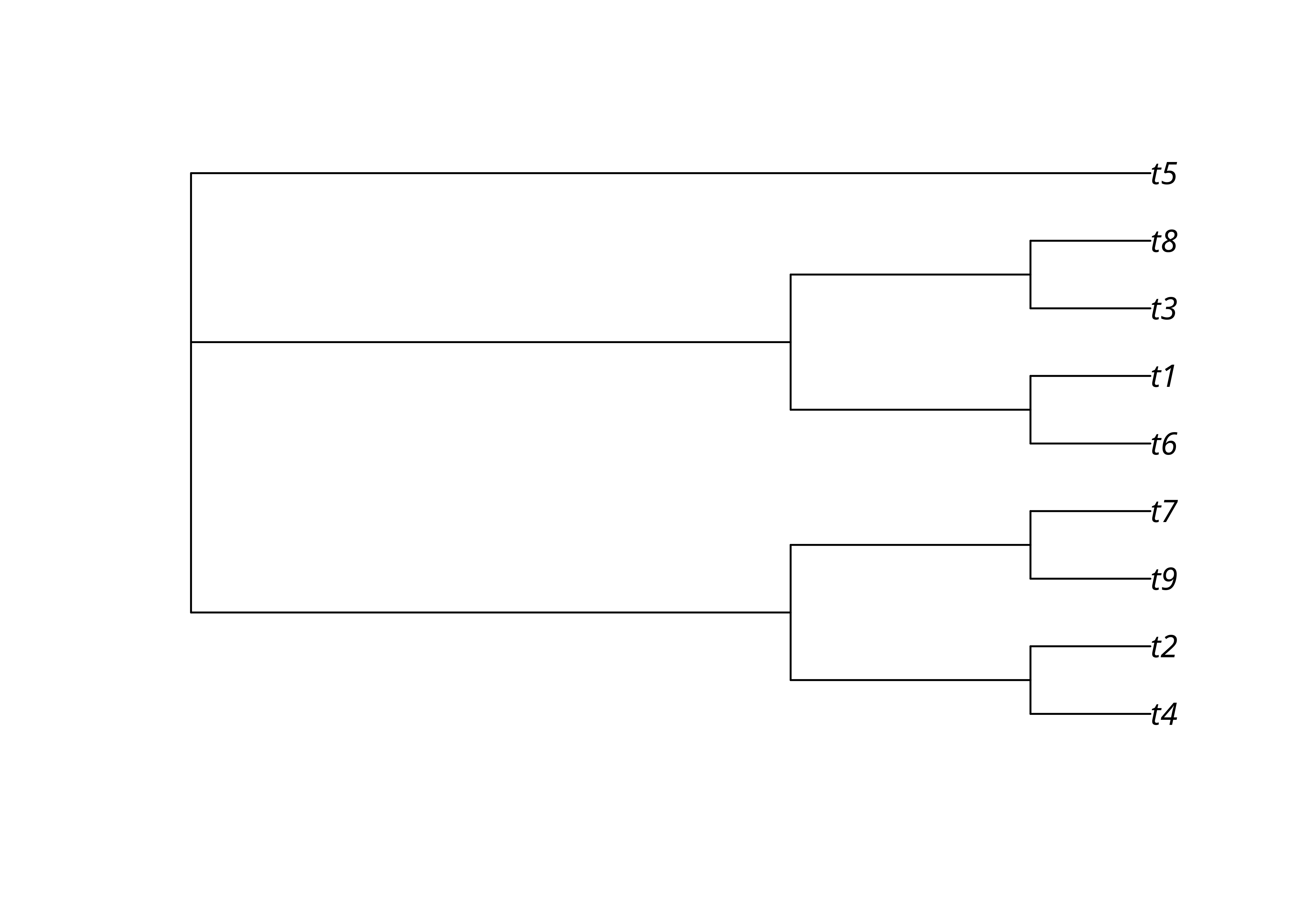
Une analyse phylogénétique résulte souvent non pas en un arbre unique mais en un ensemble d’arbres. Ce n’est le cas avec les résultats de nos analyses précédentes, mais on peut générer des données aléatoires pour l’exemple.
trees <- map(1:3, ~ rtree(n = 9))
trees <- as.multiPhylo(c(trees[1], trees[1], trees[1], trees, trees))Il est possible de résumer l’accord entre ces arbres par un arbre consensus qui ne contient que les clades présents dans une proportion p des arbres. Un arbre consensus est donc normalement moins résolu que les arbres obtenus.
Un arbre consensus strict ne contient que les clades présents dans tous les arbres (p = 1), ce qui peut parfois aboutir à un «râteau» sans aucun clade à l’intérieur de l’arbre comme ici avec nos données aléatoires.
Un arbre consensus n’a pas en principe de longueurs de branches.

Un arbre consensus majoritaire contient lui les clades présents dans la majorité (p ≥ .5) des arbres.
4.3 Mesures du degré d’arborescence
Il existe plusieurs mesures du degré d’arborescence de données phylogénétiques indiquant à quel point les données sont compatibles avec un arbre (quel qu’il soit).
4.3.1 Score δ
Le score δ est une mesure du degré de conflit d’un jeu de données. Chaque taxon se voit attribué un score entre 0 et 1 en fonction de son degré d’implication dans les signaux contradictoires. Le score δ pour un taxon se base sur la distance entre paires de taxons à l’intérieur de tous les quartets (sous-ensembles de quatre taxons) incluant le taxon en question.
Soit un quartet q composé de quatre taxons i, j ,k, l et les sommes des distance \(d_{ij} + d_{kl}\), \(d_{ik} + d_{il}\) et \(d_{il} + d_{jk}\). Soit \(m_1\) la plus grande de ces trois sommes, \(m_2\) la seconde, et \(m_3\) la plus petite. Le score δ du quartet q est égal au rapport entre les différences de ces sommes:
\[\delta_q = \frac{m_1 - m_2}{m_1 - m_3}, \delta_q = 0\ \text{si}\ m_1 - m_3 = 0\]
Le score δ d’un taxon est la moyenne des scores δ des quartets incluant le taxon, et le score δ d’un jeu de données est la moyenne des scores δ de tous les quartets.
La fonction delta.plot() permet de calculer le score δ d’une matrice de distance et d’afficher un histogramme des valeurs pour l’ensemble des quartets et la moyenne pour chaque taxon.
lx_delta <- lx_phy %>%
Hamming() %>%
delta.plot()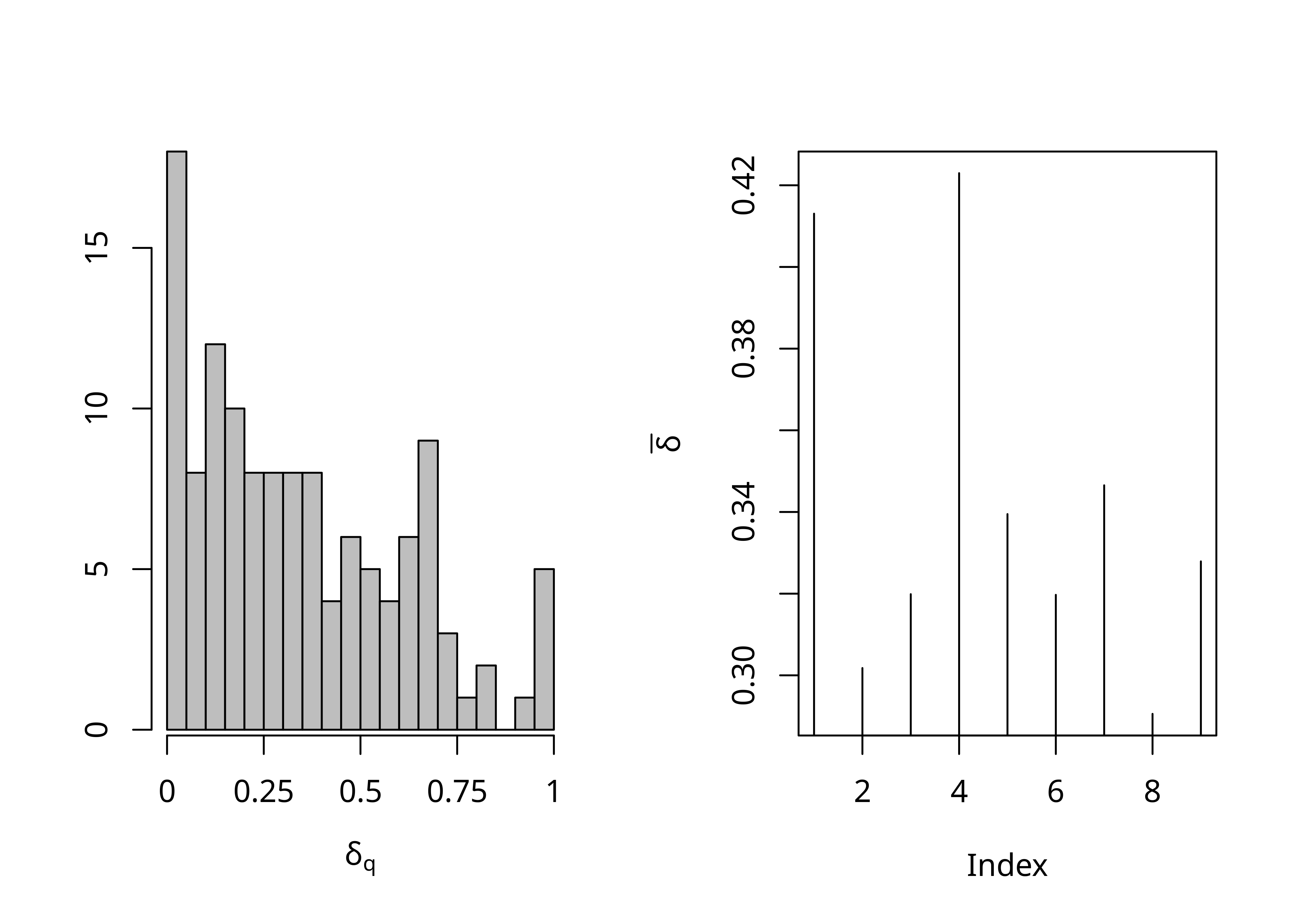
lx_delta$counts
[1] 18 8 12 10 8 8 8 8 4 6 5 4 6 9 3 1 2 0 1 5
$delta.bar
[1] 0.4130480 0.3017785 0.3198701 0.4229674 0.3394789 0.3197053 0.3465301
[8] 0.2905615 0.3279005Les moyennes de chaque taxon sont stockées dans l’attribut delta.bar, ce qui permet de calculer la moyenne.
lx_delta$delta.bar[1] 0.4130480 0.3017785 0.3198701 0.4229674 0.3394789 0.3197053 0.3465301
[8] 0.2905615 0.3279005mean(lx_delta$delta.bar)[1] 0.3424267Il n’est cependant pas possible de récupérer l’ensemble des scores pour les différents quartets. Il faut pour cela utiliser la fonction delta.score() de phangorn, qui a le désavantage d’utiliser dist.hamming() en interne à la place de Hamming(). On peut cependant copier le code de la fonction delta.score() pour créer une nouvelle fonction prennant en entrée une matrice de données et laissant le choix de la fonction de calcul du score en prévision de la suite.
delta.score2 <- function(x, arg = "mean", f = phangorn:::delta.quartet) {
dist.dna <- as.matrix(x)
all.quartets <- t(combn(attributes(x)$Labels, 4))
delta.values <- apply(
all.quartets[, ], 1, f,
dist.dna
)
if (!arg %in% c("all", "mean", "sd")) {
stop("return options are: all, mean, or sd")
}
if (arg == "all") {
return(delta.values)
}
if (arg == "mean") {
return(mean(delta.values))
}
if (arg == "sd") {
return(sd(delta.values))
}
}On peut alors obtenir l’ensemble des score δ et tracer son propre diagramme de distribution.
[1] 3.883439e-02 2.682001e-01 6.545383e-02 2.310903e-01 1.649283e-01
[6] 1.678621e-01 9.566356e-01 5.664919e-01 8.387498e-01 3.829895e-01
[11] 6.115951e-01 7.196502e-01 4.318928e-01 6.563305e-01 2.435892e-01
[16] 9.577462e-01 1.716737e-01 2.297055e-01 1.647239e-02 4.304405e-03
[21] 4.527916e-03 9.348442e-01 6.018237e-01 6.826347e-01 4.682604e-01
[26] 6.990909e-01 1.000000e+00 2.467532e-02 4.893112e-01 7.355769e-01
[31] 6.692073e-01 1.099290e-01 1.444444e-01 6.645903e-01 9.908884e-01
[36] 2.199776e-03 3.273810e-01 1.586998e-01 1.199095e-01 5.541562e-01
[41] 3.689884e-01 6.690439e-01 6.450617e-01 6.696596e-03 3.388805e-01
[46] 1.000000e+00 2.045455e-01 3.267477e-01 1.096710e-01 3.730769e-01
[51] 1.511364e-01 4.088146e-01 2.849576e-01 2.025665e-01 7.541187e-01
[56] 1.800362e-01 2.925265e-01 5.288462e-02 2.387387e-01 1.031623e-01
[61] 1.415584e-01 8.880471e-02 2.501828e-01 8.520990e-03 1.052131e-02
[66] 2.254902e-01 3.428819e-02 4.125286e-02 2.593174e-01 3.011204e-01
[71] 1.106246e-03 3.829930e-01 3.455388e-01 4.980067e-01 1.398278e-01
[76] 3.974122e-01 1.021876e-01 6.011132e-01 5.539291e-01 7.419355e-01
[81] 5.017955e-01 5.156927e-01 6.462147e-02 2.502278e-01 6.989878e-01
[86] 6.341198e-01 3.128813e-01 7.434773e-02 9.075630e-02 3.070546e-02
[91] 1.843180e-01 5.019011e-01 1.164384e-01 6.671156e-01 4.272727e-01
[96] 3.846154e-01 4.524887e-03 4.545455e-01 5.386779e-01 6.250000e-01
[101] 4.169826e-01 6.555891e-01 1.253367e-01 2.018293e-01 5.078659e-01
[106] 2.561798e-01 3.134049e-01 1.117855e-01 4.361590e-16 2.660754e-01
[111] 1.342649e-01 6.986634e-02 4.899832e-01 3.970315e-01 8.013468e-02
[116] 1.640091e-01 3.619529e-03 3.891892e-01 8.333333e-01 4.626168e-01
[121] 5.949367e-01 2.178175e-04 1.679351e-01 0.000000e+00 1.767875e-01
[126] 3.051360e-01delta.score2(lx_dist, "mean")[1] 0.3424267[1] 0.3424267delta.score2(lx_dist, "all") %>%
enframe() %>%
ggplot(aes(x = value)) +
geom_density(fill = few_pal("Medium")(2)[1], color = few_pal("Dark")(2)[1]) +
xlab("δ") +
theme_minimal()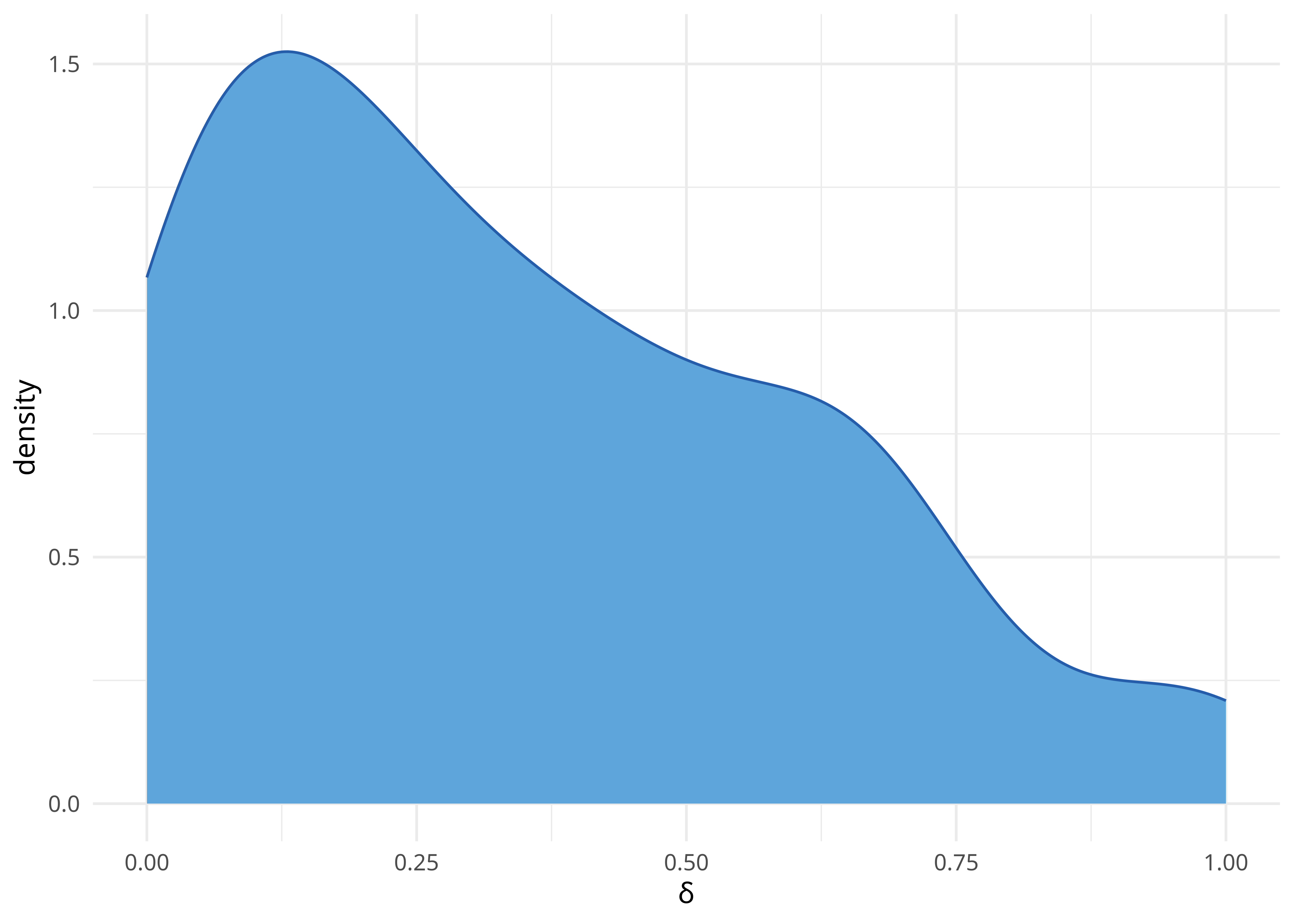
4.3.2 Score de résidus Q
Un score alternatif calculé comme \((m_1 - m_2)^2\) a également été proposé. On peut reprendre le code de la fonction cachée phangorn:::delta.quartet pour définir une nouvelle fonction à passer à notre delta.score2(). Il est nécessaire de mettre au préalable les distances à l’échelle pour que leur moyenne soit égale à 1.
qresid.score <- function(quartet, dist.dna) {
m1 <- dist.dna[quartet[1], quartet[2]] + dist.dna[
quartet[3],
quartet[4]
]
m2 <- dist.dna[quartet[1], quartet[3]] + dist.dna[
quartet[2],
quartet[4]
]
m3 <- dist.dna[quartet[1], quartet[4]] + dist.dna[
quartet[2],
quartet[3]
]
m <- sort(c(m1, m2, m3), decreasing = TRUE)
ret <- (m[1] - m[2])^2
return(ret)
}
lx_dist2 <- lx_dist/mean(lx_dist)
delta.score2(lx_dist2, "mean", f = qresid.score)[1] 0.02797994delta.score2(lx_dist2, "all", f = qresid.score) %>%
enframe() %>%
ggplot(aes(x = value)) +
geom_density(fill = few_pal("Medium")(2)[1], color = few_pal("Dark")(2)[1]) +
xlab("Q-residual") +
theme_minimal()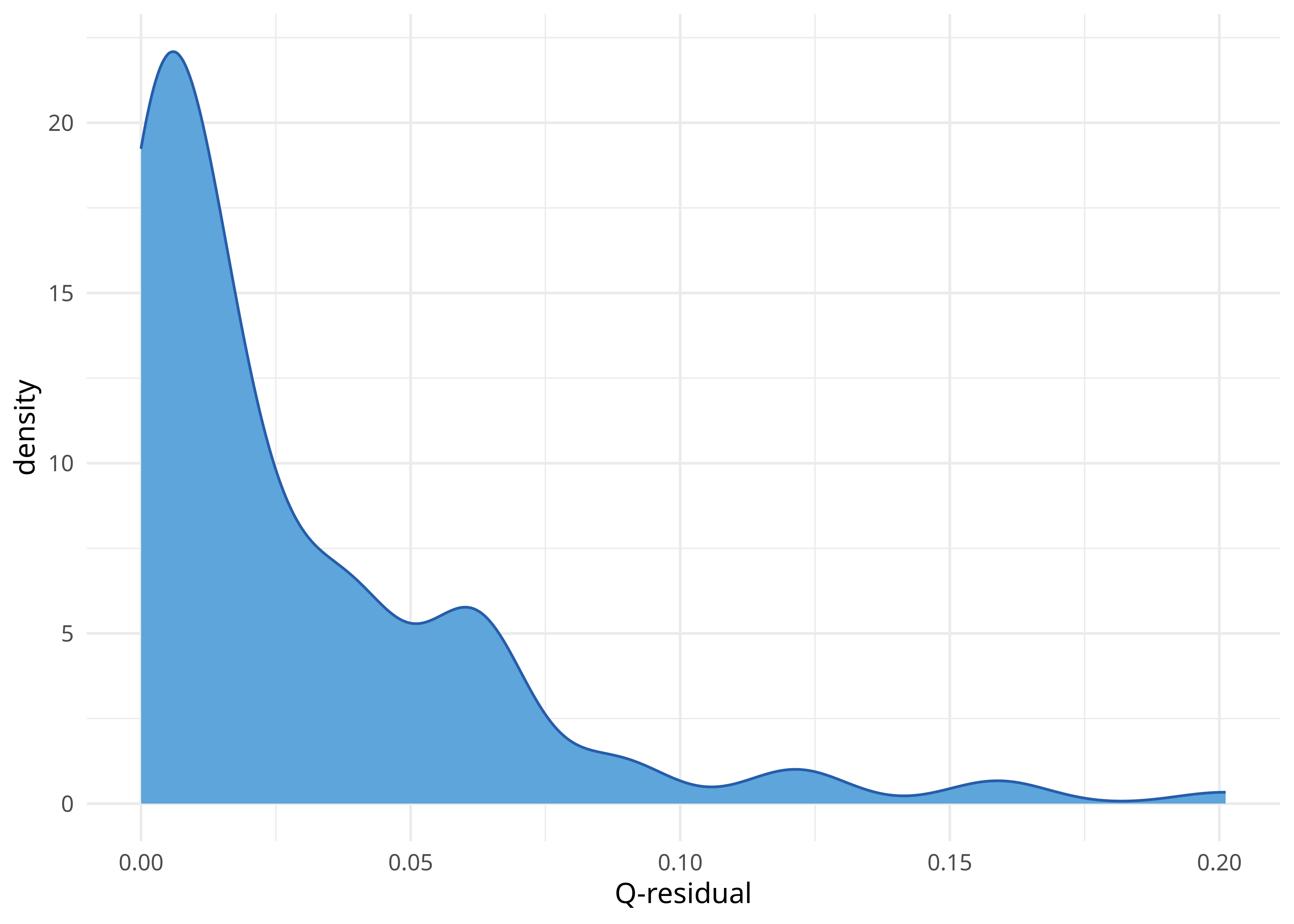
On peut ensuite comparer les scores δ et les scores de résidus Q.
ds <- delta.score2(lx_dist, "all")
qr <- delta.score2(lx_dist2, "all", f = qresid.score)
tibble(ds, qr) %>%
ggplot(aes(x = ds, y = qr)) +
geom_vline(xintercept = mean(ds), linewidth = .4) +
geom_hline(yintercept = mean(qr), linewidth = .4) +
geom_point(color = few_pal("Dark")(2)[2], fill = few_pal("Dark")(2)[2], pch = 21, alpha = .5, size = 4) +
xlab("δ") +
ylab("Q-residual") +
theme_minimal()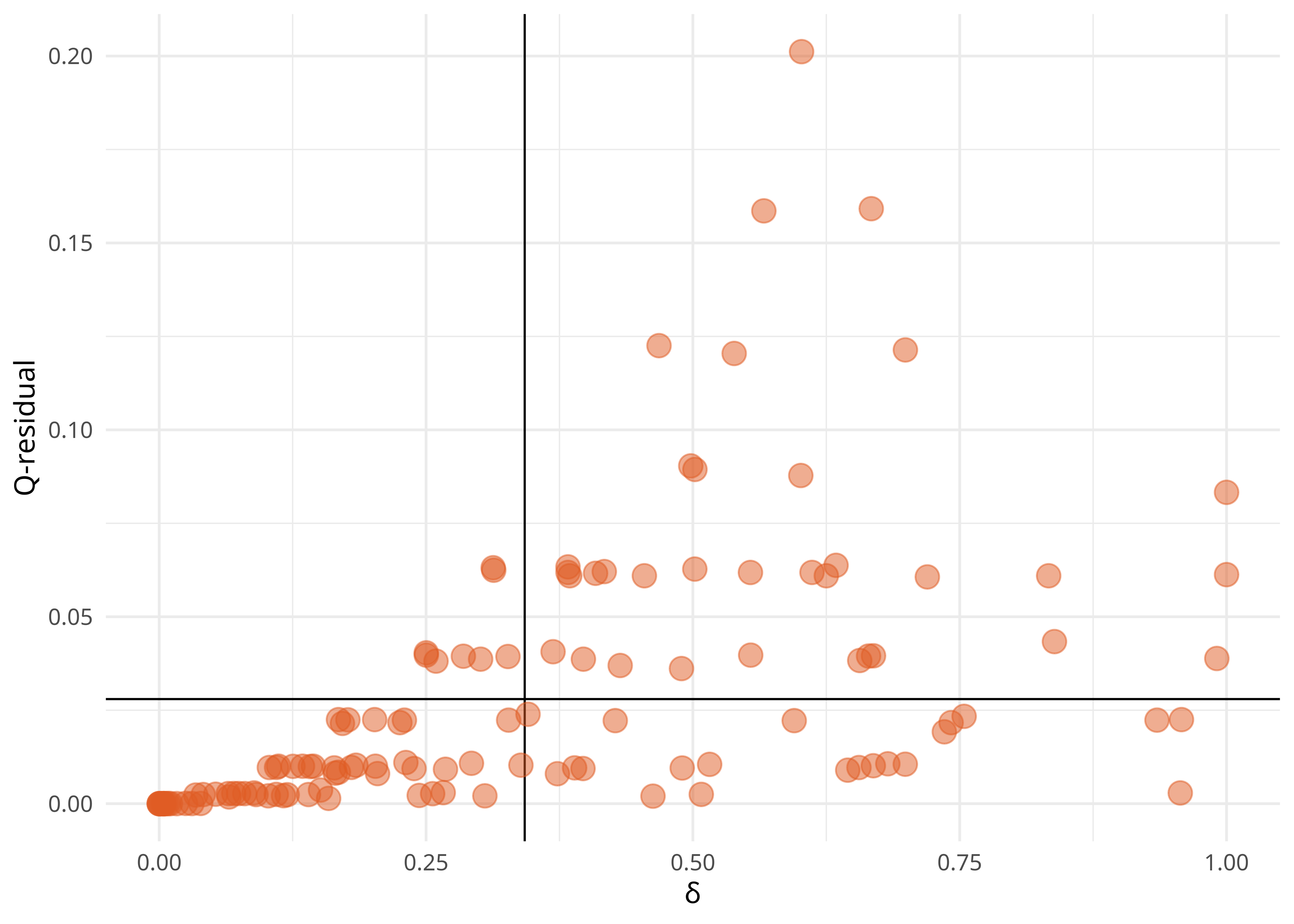
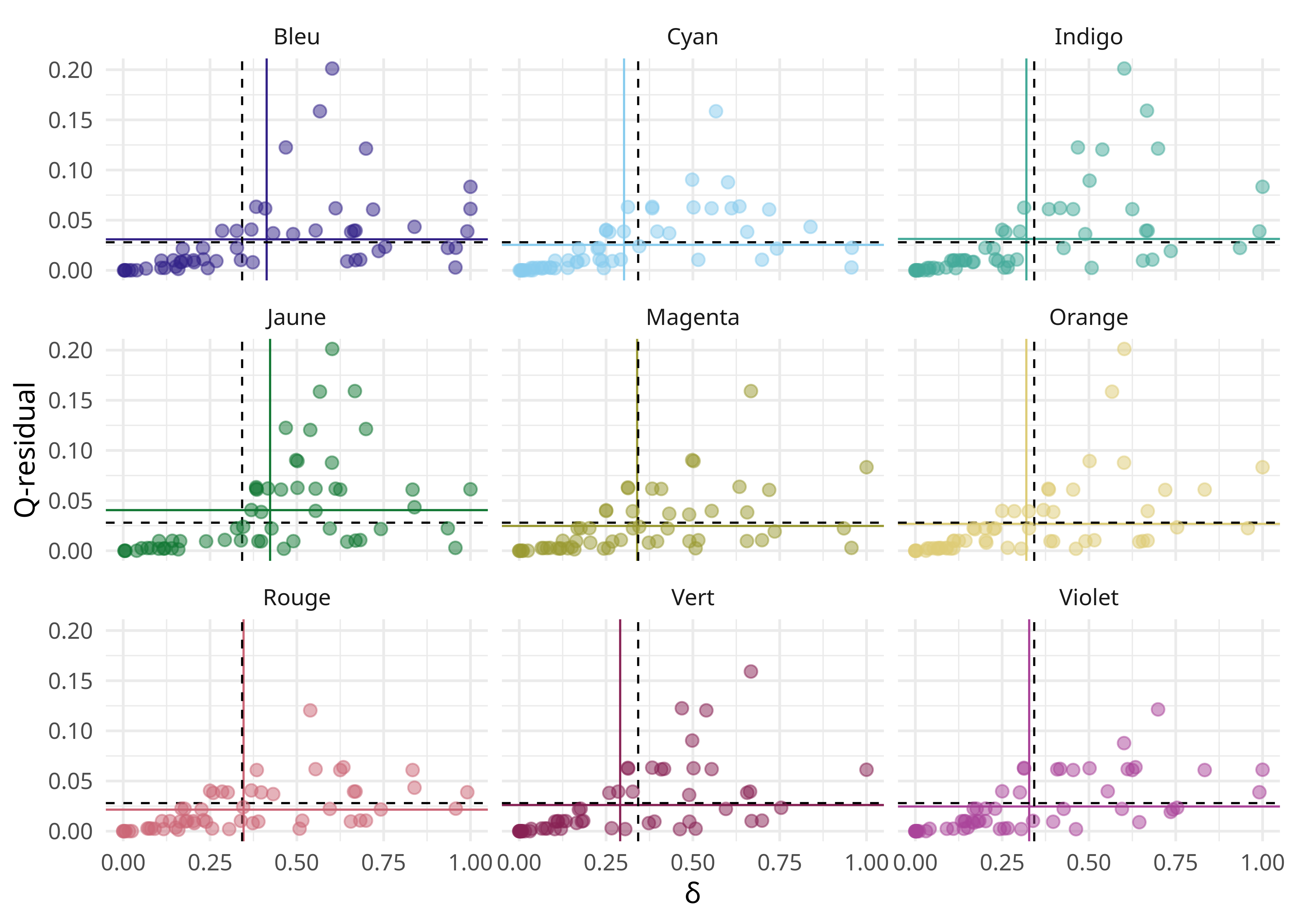
Les score δ et de résidus Q globaux sont difficiles à interpréter, mais il est néanmoins possible d’obtenir les scores pour chaque taxon et de comparer ces scores entre taxons. On constate que c’est le taxon Jaune qui a les plus mauvais scores et qui est donc le plus problématique.
dsqr_bytaxon %>%
ggplot(aes(x = ds, y = qr, color = taxon, fill = taxon)) +
geom_vline(data = dsqr_bytaxon_mean, aes(xintercept = ds, color = taxon), linewidth = .4) +
geom_hline(data = dsqr_bytaxon_mean, aes(yintercept = qr, color = taxon), linewidth = .4) +
geom_vline(xintercept = mean(ds), linewidth = .4, linetype = "dashed") +
geom_hline(yintercept = mean(qr), linewidth = .4, linetype = "dashed") +
geom_point(pch = 21, alpha = .5, size = 2) +
xlab("δ") +
ylab("Q-residual") +
facet_wrap(~taxon) +
scale_color_ptol() +
scale_fill_ptol() +
theme_minimal() +
theme(legend.position = "none")